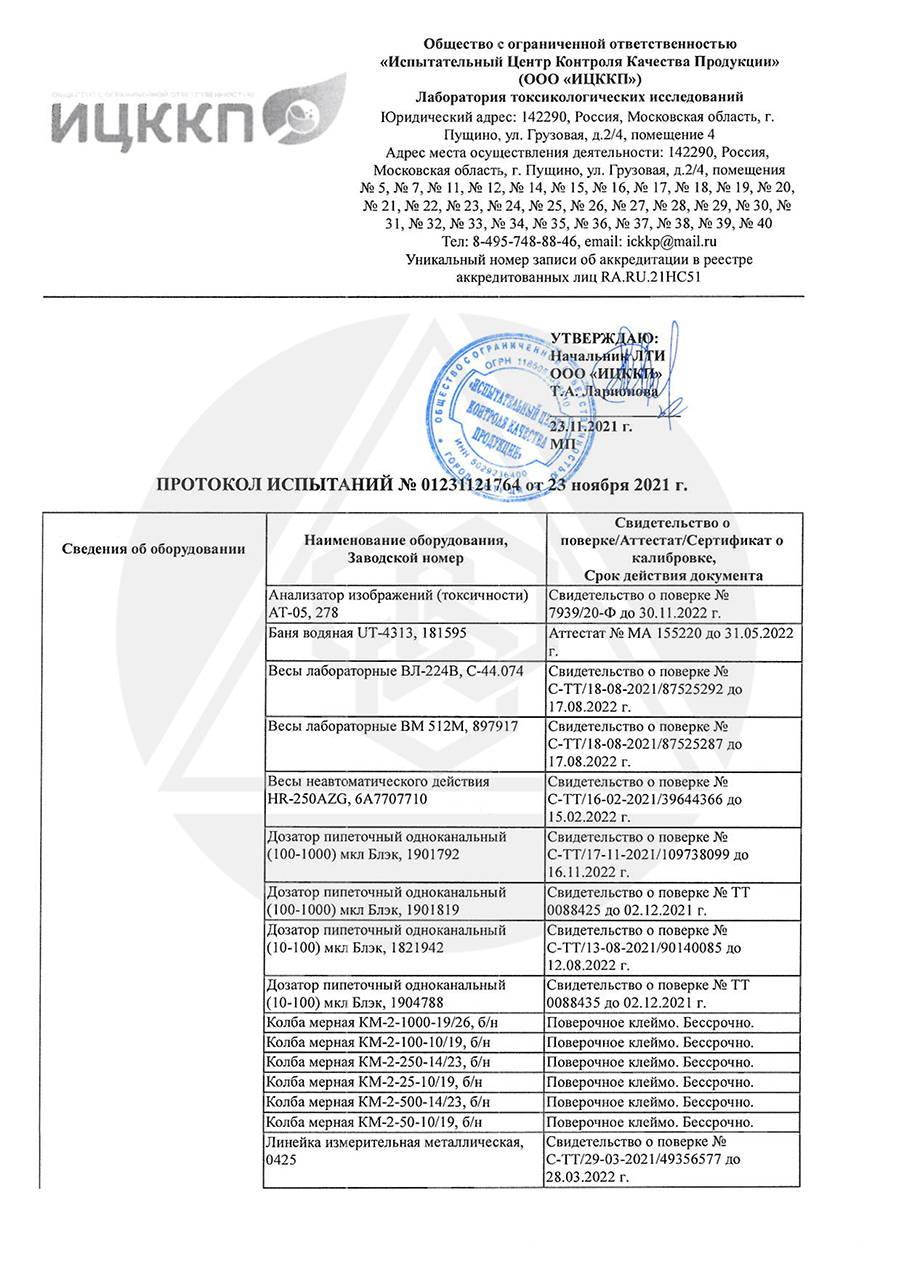
Ввод в обращение медицинского изделия (МИ) на территории РФ включает в себя следующие этапы:
- Технические испытания
- Токсикологические исследования
- Подготовка регистрационного досье на МИ с проведением I этапа экспертизы качества, эффективности и безопасности
- Клинические испытания МИ
- II этап экспертизы качества, эффективности и безопасности МИ
- Государственная регистрация мед. изделие с оформлением РУ
- Производство (изготовление)/ввоз и выпуск МИ на потребительский рынок

Регистрационное удостоверение (РУ) медицинского изделия – это официальный документ, который:
- удостоверяет факт регистрации мед. изделия Федеральной службой по надзору в сфере здравоохранения РФ (Росздравнадзором);
- разрешает свободный ввоз, производство, распространение и применение зарегистрированного мед. изделия на территории РФ;
- подтверждает факт внесения данного мед. изделия в Государственный реестр медицинских изделий Росздравнадзора.
РУ на мед. изделия выдаёт Росздравнадзор.
Какие документы необходимо для государственной регистрации мед. изделия
Для государственной регистрации мед. изделия Заявитель (производитель/изготовитель, уполномоченный представитель изготовителя, импортёр) должен доставить в Росздравнадзор следующие документы:
- Составленное в установленной форме Заявление о государственной регистрации МИ
- Документы, подтверждающие полномочия Заявителя и принадлежность МИ Заявителю, осуществляющему на законных основаниях их ввоз с целью государственной регистрации
- Нормативная,техническая и эксплуатационная документация на мед. изделие
- Фото (размером не менее 18х24 см) общего вида мед. изделия или интерфейса медицинского ПО
- Протоколы/Акты технических испытаний
- Протоколы/Заключения токсикологических испытаний
- Протоколы (акты, заключения) клинических испытаний по типовой программе (если такие проводились)
- Для импортируемых МИ: копии РУ и/или других документов, подтверждающих факт регистрации мед. изделия в стране-производителе
- Для импортируемых МИ: реквизиты Разрешения на ввоз образцов на территорию РФ для прохождения государственной регистрации
- Документы, подтверждающие возможность производителя изготавливать МИ (сертификаты СМК ГОСТ ИСО 13485 или др.)
- Другие имеющиеся у Заявителя документы, доказывающие качество, эффективность и безопасность заявленных МИ
Нормативная база

- № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» от 21.11.2011 г.
- Постановление Правительства РФ № 1416 «Об утверждении правил регистрации медицинских изделий» от 27.12.2012 г.
- Приказ № 2н от 09.01.2014 г. «Об утверждении порядка оценки соответствия медицинских изделий в форме технических испытаний, токсикологических исследований, клинических испытаний в целях государственной регистрации медицинских изделий»
- Приказ № 89н от 15.18.2012 г. «Об утверждении Порядка проведения испытаний в целях утверждения типа средств измерений, а также перечня медицинских изделий, относящихся к средствам измерений в сфере государственного регулирования обеспечения единства измерений, в отношении которых проводятся испытания в целях утверждения типа средств измерений»
- Приказ № 11н от 19.01.2017 г. «Об утверждении требований к содержанию технической и эксплуатационной документации производителя (изготовителя) медицинских изделий»
- Решение ЕЭК от 12 февраля 2016 года № 46 "О Правилах регистрации и экспертизы безопасности, качества и эффективности медицинских изделий"
- Постановление Правительства Российской Федерации от 01.04.2022 г. № 552 «Об утверждении особенностей обращения, включая особенности государственной регистрации, медицинских изделий в случае их дефектуры или риска возникновения дефектуры в связи с введением в отношении Российской Федерации ограничительных мер экономического характера»
Какая продукция считается "Медицинским изделием"
Согласно части 1 статьи 38 № 323-ФЗ, медицинским изделием являются применяемые в медицинских целях отдельно или в сочетании между собой любые:
- инструменты,
- аппараты,
- приборы,
- оборудование,
- материалы,
- специальное программное обеспечение,
- и др.,
изготовленные для профилактики, диагностики, лечения и реабилитации заболеваний, проведения медицинских исследований, мониторинга болезней, фармацевтического и прочего воздействия на организм человека в медицинских целях.
Виды медицинских изделий, подлежащих государственной регистрации, перечислены в Приложении №1 Приказа Министерства здравоохранения РФ № 4н от 06.06. 2012 г.
В Приложении №2 настоящего Приказа приведён принцип определения мед. изделий по классам риска.
Виды испытаний медицинских изделий
Виды обязательных лабораторных исследований (испытаний), которые проходят все без исключения мед. изделий перед I (первым) этапом экспертизы в рамках процедуры государственной регистрации:
Технические
Токсикологические
По результатам анализа Протоколов (Актов. Заключений) испытаний, Экспертного заключения и других обязательных документов на мед. изделие, Росздравнадзор принимает Решение о целесообразности проведения клинических испытаний.
В отношении ряда медицинских изделий, в том числе 1-го (низкого) класса риска, государственная регистрация осуществляется по ускоренной процедуре – после первой экспертизы.
Как проходят технические испытания мед. изделий
Технические испытания – форма оценки соответствия мед. изделий установленным к ним требованиям эффективности, безопасности и качества
Технические испытания проводят аккредитованные лаборатории в порядке, утверждённом Минздравом РФ.
Приказ Минздрава России №2н Об утверждении Порядка проведения оценки соответствия МИ в целях госрегистрациЭтапы технических испытаний МИ:
- Подача Заявителем в испытательную лабораторию необходимых документов, сведений и материалов:
Заявление (установленного образца) о проведении тех. испытаний
Сведения о нормативной документации на заявленное изделие
Перечень стандартов и требований, которых соответствует заявленное на испытания медицинское изделие
Эксплуатационная и техническая документация на МИ
Конструкторская документация (чертежи, схемы и т.п.)
Фотоизображение общего вида МИ, установленного размера
Образцы мед. изделий
Материалы (спец. оборудование, ПО или др.), необходимые для проведения технических испытаний
При наличии: копии Протоколов (Актов, Заключений) проводимых ранее испытаний, сведения об опасности МИ и принятых мерах снижения остаточных рисков, сведения о клиническом применении МИ за пределами РФ, другие документы от производителя, сопровождающие конкретное мед. изделие - Анализ технической документации
- Идентификация МИ с определением класса потенциального риска и номенклатуры
- Согласование с Заявителем индивидуальной Программы тех. испытаний (при их проведении)
- Оценка соответствия МИ требованиям применимых стандартов, нормативной и эксплуатационно-технической документации
- Испытания образцов МИ по составленной Программе (при проведении испытаний)
- Определение качества и безопасности применения МИ
- Анализ полученных данных
- Доработка эксплуатационно-технической документации (при необходимости)
- Оформление и выдача Заказчику Протоколов испытаний и/или Акта оценки результатов технических испытаний медицинского изделия
Как проходят токсикологические испытания мед. изделий
-
Оценка соответствия медицинских изделий в форме токсикологических испытания предусмотрены для всех видов мед. изделий, так или иначе контактирующих с организмом человека
- Подача Заявителем в испытательную лабораторию следующих необходимых документов, сведений и материалов:
Заявление (установленного образца) о проведении токсикологических испытаний
Сведения о нормативной документации на заявленное изделие
Перечень стандартов и требований, которым соответствует конкретное медицинское изделие
Техническая, конструкторская и эксплуатационная и документация на заявленное МИ
Имеющиеся документы на материалы, субстанции, комплектующие
Для лекарственных средств (в случае их применения в составе мед. изделия): № фармакопейной статьи или № нормативной документации на фарм. субстанцию или на лекарственный препарат, включенную(ый) в государственный Реестр лекарственных средств
Копии Протоколов (Актов, Заключений) ранее проводимых испытаний, других имеющихся документов, характеризующих безопасность и качество заявленного МИ
Образцы МИ - Идентификация МИ
- Классификация МИ (определения класса риска и номенклатуры)
- Анализ доставленной документации
- Согласование с Заявителем индивидуальной Программы токсикологических испытаний
- Осуществление лабораторных исследований образцов
- Анализ и оформление результатов испытаний
- Выдача Заявителю отчёта о проведённых исследованиях в форме Протоколов и/или Заключения по результатам токсикологических исследований медицинского изделия
Что проверяют эксперты ООО "ЦСД" в процессе испытаний мед. изделий

Ключевые моменту проверки мед. изделия в рамках лабораторных испытаний:
- Оценка соответствия требованиям применимых стандартов и нормативной документации
- Оценка соответствия методов исследования и контролируемых характеристик требованиям конструкторской и эксплуатационно-технической документации производителя
- Оценка безопасности применения
В ходе технических испытаний проверяются показатели:
- физико-механические характеристики
- электромагнитная совместимость
- электробезопасность
В ходе токсикологических исследований проверяются показатели:
- Физико-химические
- Санитарно-химические
- Биологические (в условиях in vitro и in vivo)
Окончательный объём испытаний и контролируемых характеристик выбирается индивидуально для каждого вида мед. изделий.
Свяжитесь с экспертами нашего Центра и получите бесплатную консультацию по всем вопросам испытаний конкретного вида мед. изделия для целей прохождения гос. регистрации
Что надо делать Заявителю после испытаний мед. изделий
После получения положительных Актов, Заключений и/или Протоколов испытаний Заявитель:
- комплектует ими пакет обязательных документов Какие документы необходимо для государственной регистрации мед. изделия
- доставляет собранные документы в региональное подразделение Росздравнадзора;
- подаёт Заявление на гос. регистрацию своих МИ.
После рассмотрения Заявления и проверки документов Росздравнадзор формирует на заявленную продукцию регистрационное досье и отправляет его в уполномоченную экспертную организацию на первую экспертизу.
Дальнейших порядок действий определяется Росздравнадзором по результатам полученного Экспертного заключения.
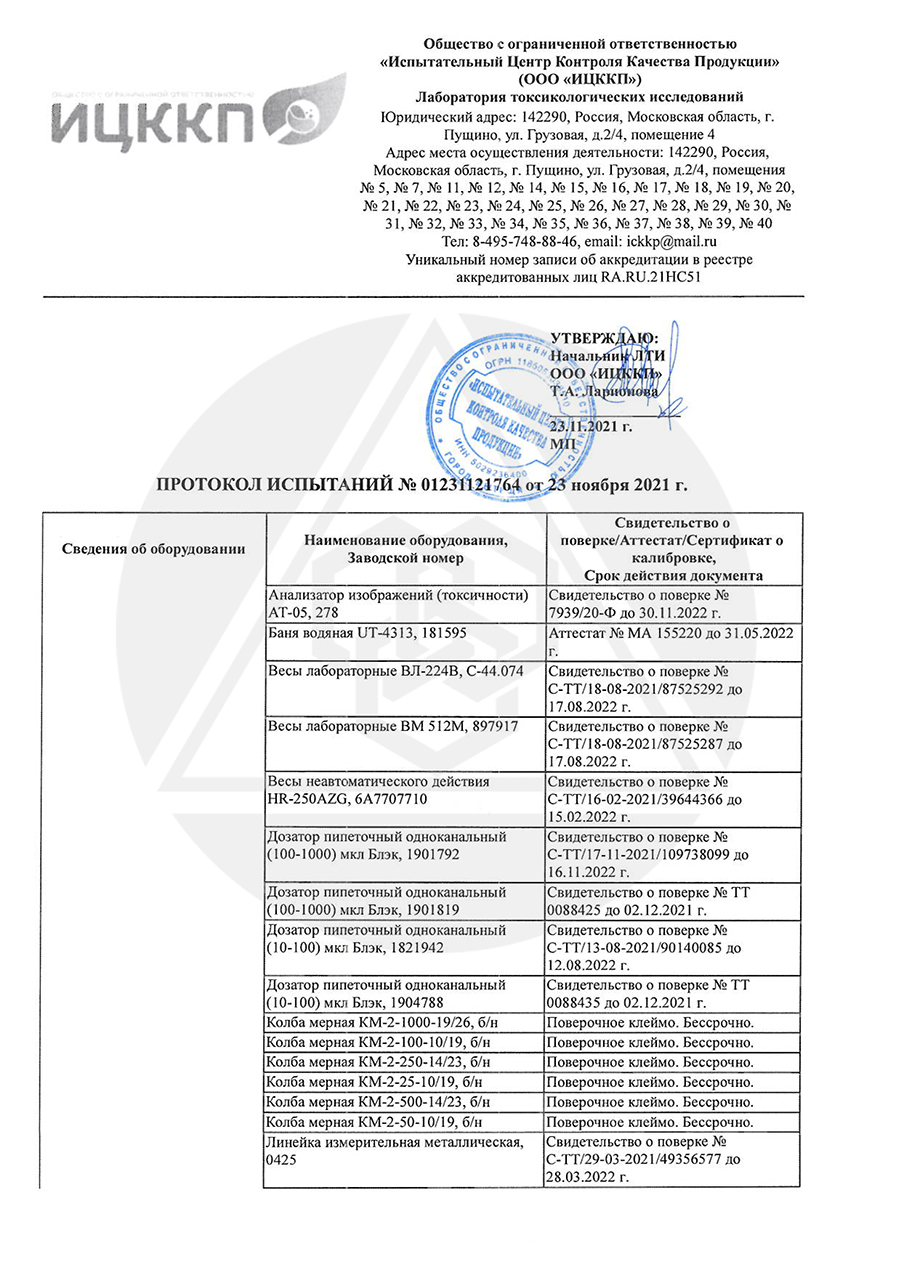
Для получения протокола испытаний требуется
Учредительные документы,
Реквизиты заявителя,
Заявление,
Техническая документация